Blood, sweat and cures
-
- from Shaastra :: vol 04 issue 01 :: Feb 2025
A recent gene therapy treatment, based on decades of research, shows promise against thalassemia and sickle cell disease.

former Principal Scientific Adviser to the Government of India, is emeritus professor at the National Centre for Biological Sciences, Tata Institute of Fundamental Research.
What do Amitabh Bachchan and Jackie Shroff have in common – other than the fact that they are actors? Both of them have thalassemia minor, a blood disorder; they have a form of the condition with minor or no symptoms, and are brand ambassadors for wider awareness about the disorder. And what do the tribal populations of Gujarat, Chhattisgarh and Tamil Nadu have in common with the late jazz musician Miles Davis and model Bethany Chasteen? Tribal populations in India, many African populations and African Americans have a high frequency of sickle cell disease, which is another blood disorder. Davis and Chasteen have it, too. And a recent gene therapy treatment can treat both thalassemia and sickle cell disease.
This shared treatment stands on several decades of research into the two diseases, and points to how long and fraught the route to disease treatment is. It also points to the complexities of so-called 'monogenic' diseases — those caused by mutations in just one gene — and how simplifying features can unravel some of this complexity to advance both understanding and solutions.
PAULING'S CONTRIBUTION
Much of the research on understanding sickle cell disease was stimulated by Linus Pauling. Pauling was a prolific, high-quality researcher. His book, The Nature of the Chemical Bond, written in 1939, when he was 38 years old, was a 'Rosetta Stone' for researchers, translating the mysteries of how atoms and molecules interact into an understanding of the chemical world. For researchers and students trying to decipher the structure of biological molecules, such as the genetic material deoxyribonucleic acid (DNA), and the protein haemoglobin – the oxygen carrier in our blood cells – Pauling's book and his research were invaluable. Pauling and his collaborators published a paper in 1949 after examining the blood of African Americans who had sickle cell disease. The authors pointed out that about 8% of African Americans had what is called sicklemia, or sickle cell trait. They displayed no abnormalities, but when their red blood cells were examined under the microscope and "the oxygen pressure is lowered, these cells change their forms from the normal biconcave disk to crescent, holly wreath, and other forms. This process is known as sickling."
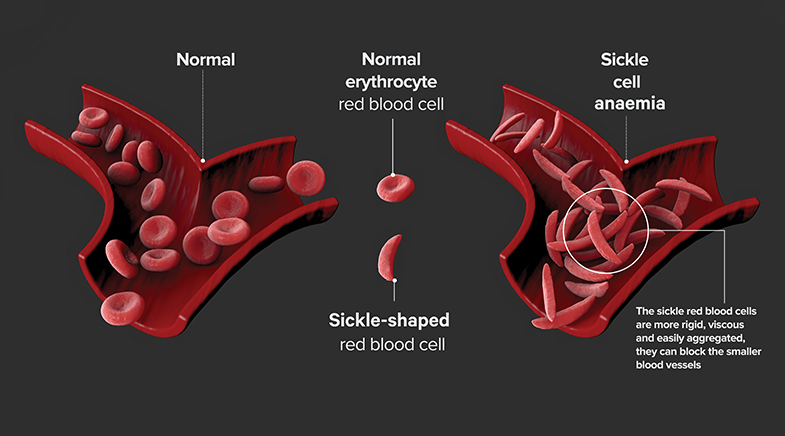
About 2% of those whose red blood cells suffer from severe anaemia have sickle cell anaemia or sickle cell disease. What was defective in the red blood cell that caused this shape change upon oxygen stress was unclear. The red blood cell contains the protein globin, to which a molecule called haem (or heme) binds. Together, they get the name haemoglobin. Heme is an organic molecule that contains iron. A defective oxygen response by haemoglobin could result from defective heme synthesis and an inability to bind iron well. Alternatively, it could result from a defect in the globin protein. Pauling and colleagues isolated haemoglobin from normal individuals, those with sickle cell trait, and those with sickle cell disease. Analysing these preparations, they examined how the haemoglobins moved when placed under the influence of an electric field. If the globin proteins from sickling red cells were defective, their surface electric charges would vary, and their mobility would differ from normal haemoglobin. Indeed, that is what they observed.
A monthly column that explores themes on nature, nurture and neighbourhood in the shaping of form and function.
Combining this conclusion with those of previous researchers on the inheritance of sickle cell traits, they concluded that a genetic mutation led to a defective globin protein. When two mutant copies were present in a person, the more severe sickle cell disease was seen. This simple conclusion was a breakthrough linking genes to polypeptides. (Polypeptides comprise a chain of amino acids. Many proteins, such as globins, can be made of a combination of identical or distinct polypeptides.) But what exactly was the nature of the defect in the haemoglobin in sickle cell disease?
INGRAM'S INTERVENTION
Vernon Ingram, considered the "father of molecular medicine", solved that riddle. In 1956, he identified the exact structural alteration in the haemoglobin protein that causes sickle cell anaemia. He discovered that the disease results from a single amino acid substitution in the beta-globin chain of haemoglobin, where one amino acid (valine) replaces another (glutamic acid) at the sixth position. This finding was revolutionary because it demonstrated for the first time that a single amino acid change could cause a genetic disease. It established sickle cell anaemia as the first "molecular disease". It provided evidence for the direct relationship between genes and protein structure. Ingram died in 2006, much recognised, but not with a Nobel, illustrating yet again how much impactful work in medicine or physiology is not awarded this prize and why we should not use the Nobel as an exclusive metric of high excellence.

Like sickle cell disease, thalassemia is an inherited blood disorder. But while defects in haemoglobin cause sickle cell disease, thalassemia is typically caused by mutations affecting the production of haemoglobin. Haemoglobin in adults typically has four polypeptide chains: two 'alpha' chains and two 'beta' chains. For the normal function of haemoglobin, they must be present in the correct ratio and the right amounts. Put simply, genes have two parts. One part is the DNA that codes for the polypeptide, as in the case of sickle cell disease. The other part is the control element that regulates the expression levels of the polypeptide coding part. A defect in the coding region leads to a defective globin polypeptide, part of the haemoglobin protein complex of four polypeptides. This is what happens in sickle cell disease. In thalassemia, defects in the control element can reduce the levels of one polypeptide chain — alpha or beta — that constitutes haemoglobin, skewing the ratio between the polypeptide sub-units that make haemoglobin. (Some thalassemias can also have mutations that cause defective proteins.) Thus, alpha-thalassemia is caused by defects in alpha globin genes. Mutations in the beta globin gene cause beta-thalassemia. These mutations lead to reduced or absent alpha or beta globin chain production. This results in an imbalance between alpha and beta globin chains. Excess unpaired globin chains precipitate in red blood cell precursors, causing ineffective red blood cell development.

For researchers and students trying to decipher the structure of biological molecules, Pauling's book and his research were invaluable.
ROAD TO A THERAPY
A way to treat both diseases with one method emerged from decades of work by stem cell biologist Stuart Orkin and many colleagues. They started by observing that some people did not show severe forms of disease even though they carried the same genetic defect in globin gene regulation as others. Such effects can happen due to 'modifiers', or mutations in other genes that can modify gene expression. One modifier of thalassemia and sickle cell disease affects the expression of foetal haemoglobin. Expressed early in development, foetal haemoglobin is 'shut down' in the adult, and the adult globin genes are 'switched on'. If foetal haemoglobin expression is not shut down in the adult, its presence can ameliorate thalassemias and sickle cell disease. To track down how this is done, Orkin and colleagues used a laborious and complex method of sleuthing called genome-wide association studies (GWAS). They asked what region of the human genome was associated with continued foetal haemoglobin expression.

Haematologist Swee Lay Thein, Orkin and their collaborators showed that a defect in the regulatory region of a gene called BCL11A was associated with continued foetal haemoglobin expression; this mutation prevents the expression of BCL11A protein in the adult, where it usually acts as a repressor foetal haemoglobin gene expression. Thus, removing a repressor of foetal haemoglobin allows for its continued expression, ameliorating adult red blood cell haemoglobin defects. They showed that the repressor works by displacing an activator. Removing the repressor allowed the activator to function and keep the foetal gene switched 'on'.
This is arduous but elegant detective work, but how could it translate to therapy? One way is to design drugs that specifically remove BCL11A in adult cells that become red blood cells. This is a difficult task in terms of both drug design and precise delivery. We don't want to remove BCL11A from other places where it may play an important role. Taking another approach, teams led by Orkin made a remarkable discovery. Working with laboratory mice, they began to analyse how BCL11A was regulated. Was there a way by which BCL11A could be switched off only in the adult, thereby allowing the expression of the foetal haemoglobin gene it repressed? They showed that this could be done in mice. Using a relatively new gene-editing method, CRISPR/Cas9, they could delete a small region in the control element of BCL11A. This region, they showed, was required to keep the gene 'on' in the adult, and when mutated it switched 'off' the expression of the BCL11A gene, allowing the expression of the foetal haemoglobin gene.
A way to treat both diseases with one method emerged from decades of work by biologists.
A mouse is not human; what works there may not work in humans. Orkin's research has led to a significant breakthrough in treating blood disorders like sickle cell disease and beta-thalassemia. His work is considered the most critical advance in this field in over 70 years. These treatments have shown promising results in clinical trials and have even led to the first approved CRISPR therapy for these diseases. Other companies are now using Orkin's findings to develop their treatments.
This research could help people worldwide who suffer from these blood disorders. It opens up new possibilities for treatment that weren't available before, potentially changing the lives of many patients. The costs of these treatments are currently very high and unaffordable for implementation at scale. But as with several therapies, there is hope that this will steadily and rapidly change.
COLLABORATIONS ACROSS TIME
There are also important lessons. Driven by curiosity, Pauling tried to understand a disease's chemical basis. This is an example of 'applied' research by one of the most outstanding 'basic' researchers in chemistry. Ingram's work clarified our concept of the gene. This resulted in thousands of human-years of work understanding the gene and how it is regulated. This facilitated the basic and applied work of those such as Orkin. Applications and solutions to problems through science are like an unlimited water supply from the well of knowledge. But such wells must be constantly fed by diverse streams over decades and centuries and not allowed to run dry. Work on haemoglobin therapies exemplifies this beautifully.
Also Read:
Linus Pauling et al. Sickle Cell Anemia, a Molecular Disease. Science, Vol 110, Issue 2865, 543-548 (1949). bit.ly/pauling-sickle
Ingram V.M. A Specific Chemical Difference Between the Globins of Normal Human and Sickle-Cell Anaemia Haemoglobin. Nature, 178, 792-794 (1956). go.nature.com/4jnRIyR
Daniel E. Bauer et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science, 342, 253-257 (2013). bit.ly/BCL11A-enhancer
See also:
A whole new way of looking at medicine: Siddhartha Mukherjee
A healthcare revolution is in the works
Closer to a cure










Towards a green future
Scientists are looking at cost-effective and sustainable ways of producing green hydrogen. Using wastewater is one of them.

How AI helps put down forest fires
As instances of wildfires increase, AI and data are being tapped to predict, detect and manage forest fires.

This telescope in Ooty is over the muon
CRL scientists are studying the mysteries of cosmic rays with the help of an indigenous muon telescope, the most sensitive on Earth.

Have a
story idea?
Tell us.
Do you have a recent research paper or an idea for a science/technology-themed article that you'd like to tell us about?
GET IN TOUCH